The U.S. generic drug market moves billions of prescriptions every year-over 90% of all prescriptions filled. But behind that number is a complex, costly, and tightly regulated system that depends on one key law: the Generic Drug User Fee Amendments, or GDUFA. Without it, the FDA would be stuck reviewing thousands of applications with outdated funding, leaving patients waiting months or even years for affordable medicines. GDUFA changed that. It didn’t just add money-it rebuilt how the FDA works with generic drug makers to get safe, low-cost drugs to the public faster.
What GDUFA Actually Does
GDUFA isn’t a vague policy. It’s a legally binding agreement between the FDA and generic drug manufacturers. Signed into law in 2012, it lets the FDA collect fees directly from companies that make or sell generic drugs. These aren’t taxes. They’re user fees-paid only by those who benefit from faster FDA reviews. In return, the FDA promises to meet clear deadlines: review applications within specific timeframes, inspect manufacturing sites regularly, and provide clearer guidance. Before GDUFA, the FDA had a backlog of over 3,000 pending generic drug applications. Some had been waiting more than five years. The agency was underfunded, understaffed, and overwhelmed. GDUFA gave them the resources to hire more reviewers, upgrade technology, and conduct more inspections-especially overseas. Today, the average review time for a generic drug application is under 10 months, down from over 30 months in 2012.How the Fees Work
The fee structure is detailed and tiered. Generic drug companies pay four main types of fees:- Facility fees: Paid annually by every site that makes active ingredients (API) or finished drug products (FDF). Domestic facilities pay less than foreign ones. In 2025, a U.S. finished drug facility pays $205,500; a foreign one pays $220,500.
- Application fees: A one-time charge when a company submits an Abbreviated New Drug Application (ANDA). This covers the FDA’s review of the drug’s safety, effectiveness, and manufacturing process.
- Drug Master File (DMF) fees: Paid when a supplier first references its chemical or manufacturing data in an ANDA.
- Prior Approval Supplement (PAS) fees: Charged each time a company changes its manufacturing process after approval.
Why GDUFA Was Needed
In the early 2010s, the generic drug system was broken. Companies spent years waiting for approval. Some never got there. Meanwhile, patients paid more for brand-name drugs because generics couldn’t enter the market. The FDA was forced to make decisions based on limited resources, not science. GDUFA fixed that by tying funding directly to performance. The FDA now publishes monthly reports showing how many applications they’ve reviewed, how many inspections they’ve completed, and how many are still pending. Transparency became part of the deal. And it worked. By 2017, the backlog had dropped to under 600 applications. In 2024, it’s below 300. But it wasn’t just about speed. GDUFA forced the FDA to become more predictable. Companies now know exactly when to expect feedback. They can plan product launches. Investors can fund new generics with confidence. That’s huge for a sector where timing means profit-or failure.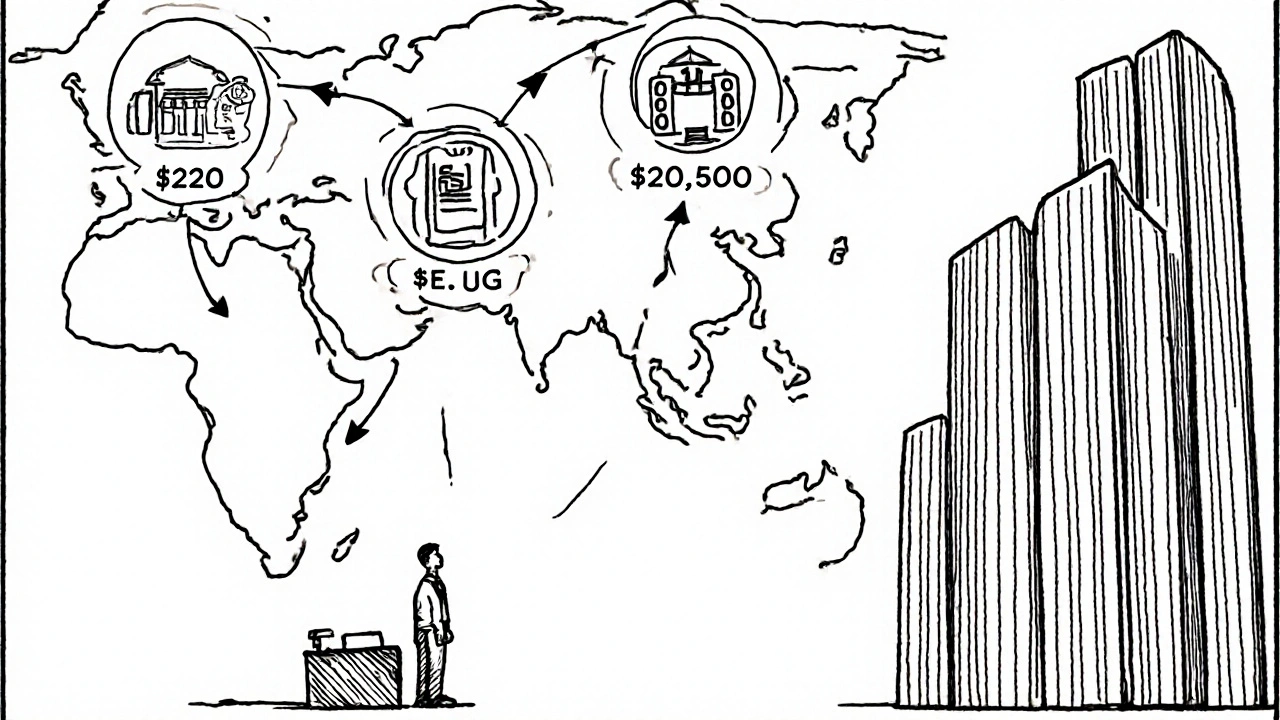
How GDUFA Changed Over Time
GDUFA isn’t static. It renews every five years. Each version fixes problems from the last.- GDUFA I (2012-2017): Built the system. Added fees. Cut the backlog. But it hurt small companies. The fixed annual facility fee was the same whether you made one drug or fifty. A startup with one product paid the same as a giant like Teva or Sandoz. That made it harder for new players to enter.
- GDUFA II (2018-2022): Fixed the small-business problem. Introduced a “small business” discount for companies with fewer than 500 employees and less than $10 million in gross revenue. Also added more flexibility for complex generics.
- GDUFA III (2023-2027): Focused on complexity. Added the Pre-ANDA Program, where companies can meet with FDA scientists before submitting a full application. This is a game-changer for drugs like inhalers, injectables, or topical creams that are hard to copy. It reduces rejection rates and saves time. Also introduced the ANDA Assessment Program, which gives early feedback on application quality.
Who Pays and Who Benefits
The biggest payers are foreign manufacturers. About 80% of generic drug ingredients and 40% of finished products in the U.S. come from overseas-mostly India and China. Those companies pay the higher facility fees. Some argue the $15,000 difference is outdated. Others say inspections abroad still cost more. On the other side, U.S.-based generic makers benefit from faster approvals and more predictable timelines. But they also face pressure. The fees are rising. In 2013, total user fees were around $300 million a year. By 2025, they’re over $500 million. That’s a big cost for companies that sell drugs for pennies. Patients win, though. Generic drugs save the U.S. healthcare system over $300 billion every year. GDUFA keeps that flow going. Without it, fewer generics would get approved. Prices would rise. Access would drop.
Challenges and Criticisms
Despite its success, GDUFA isn’t without problems. Small companies still struggle. Even with the discount, the $205,500 facility fee is a huge upfront cost. Many startups delay applying until they have multiple products ready to submit together. That slows innovation. Foreign manufacturers complain the fee structure doesn’t reflect actual inspection costs. A plant in Bangalore might be easier to inspect than one in rural Ohio, but they still pay the same premium. There’s also concern about consolidation. The top 10 generic drugmakers now control more than half the U.S. market. GDUFA’s fixed fees favor big players who can spread costs across dozens of products. Smaller firms get squeezed out. And while the FDA publishes reports, some experts say the data doesn’t always show the full picture. A “review completed” doesn’t mean the drug was approved. It might have been sent back for more data. That delay isn’t tracked the same way.What Comes Next
GDUFA III expires on September 30, 2027. Negotiations for GDUFA IV will likely start in 2026. Industry groups are already pushing for:- More fee relief for small businesses
- Lower fees for complex generics
- Digital-only submissions to cut paperwork
- More transparency on inspection outcomes
What is GDUFA and why does it matter?
GDUFA stands for Generic Drug User Fee Amendments. It’s a U.S. law that lets the FDA collect fees from generic drug manufacturers to fund the review of generic drug applications. Before GDUFA, the FDA had a massive backlog of applications, delaying access to affordable medicines. Since 2012, GDUFA has cut review times by more than 70% and increased inspections of drug factories, both domestic and overseas. It matters because it keeps low-cost generic drugs flowing to patients who need them.
Who pays the GDUFA fees?
Generic drug manufacturers pay GDUFA fees. This includes companies that make active ingredients (API), finished drug products (like pills or injections), and those who submit applications to the FDA. Foreign manufacturers pay higher fees than U.S.-based ones-$15,000 more per facility-because inspections overseas cost more. The fees are mandatory: if you want your generic drug approved in the U.S., you pay.
How much do GDUFA fees cost in 2025?
In 2025, the annual facility fee for a U.S. finished drug product site is $205,500. For a foreign site, it’s $220,500. The fee for an active pharmaceutical ingredient (API) facility is $109,000 domestically and $124,000 overseas. There’s also a one-time application fee of $1,145,000 for a new ANDA submission. These fees are set by the FDA and updated each fiscal year.
Does GDUFA help small generic drug companies?
Yes, but only partially. GDUFA II introduced a small business discount for companies with fewer than 500 employees and under $10 million in revenue. These companies pay reduced facility fees. But the fees are still high compared to revenue for startups. Many small firms wait to submit multiple applications at once to spread the cost. While GDUFA improved predictability, it didn’t fully solve the barrier to entry for new players.
What’s the difference between GDUFA I, II, and III?
GDUFA I (2012-2017) created the fee system and cleared the backlog. GDUFA II (2018-2022) added discounts for small businesses and improved transparency. GDUFA III (2023-2027) focuses on complex generics, with new programs like Pre-ANDA, which lets companies consult with FDA scientists before submitting applications. Each version built on the last, fixing flaws and expanding scope.
Is GDUFA funded by taxpayer money?
No. GDUFA is funded entirely by user fees paid by generic drug manufacturers. Congress prohibits the FDA from using general tax revenue for generic drug review under GDUFA. All fees collected must be spent only on activities related to reviewing applications, inspecting facilities, and improving the generic drug program. This ensures the industry pays for the review it receives.
What happens after GDUFA III ends in 2027?
If Congress doesn’t pass new legislation by September 30, 2027, the FDA will no longer be allowed to collect GDUFA fees. That would mean no more user fee funding for generic drug reviews. The backlog would likely return, review times would slow, and fewer generics would reach the market. Negotiations for GDUFA IV are expected to begin in 2026 to avoid this disruption.
All Comments
David Barry November 13, 2025
Let’s be real-GDUFA is just corporate welfare with a side of FDA bureaucracy. The $220K foreign facility fee? That’s a tariff in disguise. India’s manufacturing is cheaper, faster, and often better quality. Yet we punish them with inflated fees while domestic giants get cozy with the FDA. This isn’t patient protection-it’s protectionism dressed up as regulation.
Benjamin Stöffler November 14, 2025
It’s fascinating-GDUFA doesn’t just streamline review; it transforms the FDA from a reactive, underfunded relic into a performance-driven, fee-funded engine of pharmaceutical efficiency. The fee structure? A masterclass in incentive alignment. Companies pay for speed, and the FDA delivers on deadlines. It’s capitalism with public health guardrails-and yes, the foreign premium? Totally justified. Inspecting a facility in Hyderabad requires more than a plane ticket-it requires cultural fluency, language support, and logistical nightmares that U.S. sites simply don’t pose.
And yet, critics miss the forest for the trees: without this system, we’d still be waiting 3+ years for a generic version of a $10,000-a-year drug. Patients die waiting. GDUFA doesn’t just save money-it saves lives.
Also, the Pre-ANDA program? Genius. It’s like giving startups a roadmap instead of a minefield. No more blind submissions. No more six-month radio silence. Just science-driven collaboration. This isn’t bureaucracy-it’s engineering.
And the small business discount? It’s not perfect-but it’s a start. The real problem isn’t GDUFA; it’s the fact that drug development is inherently capital-intensive. The fee structure reflects reality, not malice.
Let’s not pretend this is perfect. But it’s the best damn system we’ve ever had for generic drugs. And if you think taxpayer money should fund this? You’re delusional. The industry benefits. The industry pays. Simple.
Also-why do people keep saying “the FDA is slow”? That was 2011. Today? The backlog’s under 300. We’ve cut review time by 70%. That’s not failure. That’s transformation.
And yes, the fees are rising. But so is complexity. Inhalers, injectables, biosimilars-they’re not aspirin. They require more science, more scrutiny, more time. The fee increases? They’re proportional. And if you can’t afford it? Then maybe you shouldn’t be in this space.
Bottom line: GDUFA isn’t a tax. It’s a contract. And it’s working.
Mark Rutkowski November 14, 2025
I’ve always believed that systems that align incentives with outcomes are the most beautiful kind of human engineering. GDUFA isn’t just about money-it’s about trust. The FDA says, ‘We’ll move faster if you pay fairly.’ The industry says, ‘We’ll pay if you deliver.’ And for over a decade, they’ve kept that promise. That’s rare. That’s sacred.
It’s not perfect. Small companies still sweat over fees. Foreign plants still feel the sting of that $15K premium. But look at the results: 90% of prescriptions filled with generics? That’s not luck. That’s design. That’s a quiet revolution in healthcare access.
And when GDUFA IV comes? Let’s not break what’s working. Let’s refine it. Maybe lower fees for truly small innovators. Maybe add digital-first submissions. Maybe even let startups batch applications without penalty. But don’t scrap the model. This system lets a kid in Ohio get insulin for $25 instead of $300. That’s not policy. That’s humanity.
Ryan Everhart November 16, 2025
So the FDA charges companies to review their own drugs... and people are surprised this works? I mean, if you paid me to fix your car, I’d probably fix it faster too. But hey, let’s pretend this isn’t just a fee-for-service model disguised as public health policy. Classic.
Alyssa Lopez November 17, 2025
AMERICA FIRST! Why the hell are we letting foreign labs get away with paying less? We’re the land of the free and home of the brave-our companies should be the ones getting the edge! GDUFA should be even tougher on China and India. They’re stealing our jobs and our medicine! We need tariffs on pills! #MakeGenericsGreatAgain
Alex Ramos November 17, 2025
Just wanted to say-this is one of the most underrated success stories in U.S. healthcare. I work in pharma compliance, and I’ve seen the before and after. GDUFA turned chaos into clarity. The Pre-ANDA program? Life-changing for complex generics. Also, the FDA’s public dashboards? Honestly, I didn’t think they’d actually post real-time data. They did. And it’s beautiful. 🙌
edgar popa November 19, 2025
generic drugs save billions. gdufa keeps them coming. simple.
Eve Miller November 20, 2025
It’s not just about fees-it’s about accountability. The FDA must meet deadlines. Companies must submit complete applications. No more excuses. No more delays masked as ‘review.’ This is governance with teeth. And it works. The data proves it. Anyone who complains about cost is ignoring the alternative: patients waiting years for life-saving medications.
Chrisna Bronkhorst November 22, 2025
Foreign facility fees are a scam. A plant in Pune might be cleaner than a plant in rural Ohio. The cost differential isn’t $15K-it’s $1.5K max. This is protectionism disguised as cost recovery. The FDA’s audit logs don’t even show that. It’s a numbers game to justify bigger budgets. And it’s working. Too well.
Amie Wilde November 22, 2025
My cousin works at a small generic maker. They waited 2 years to submit their first ANDA because they had to save up for the fee. It’s brutal. GDUFA helped-but it didn’t fix the startup trap. We need tiered fees based on revenue, not just employee count. Just saying.
Gary Hattis November 23, 2025
From South Africa to the U.S.-this is the model every developing country should copy. No one talks about how GDUFA created a global standard. India’s generic industry didn’t just survive-it thrived because they had to meet U.S. standards. That’s not exploitation. That’s capacity-building. And now, their drugs are trusted worldwide. GDUFA didn’t just speed up reviews-it raised the bar for global pharma.
Also, the fact that the FDA publishes inspection results? That’s transparency most governments dream of. We should export this model. Not just the fees-the culture of accountability.
Esperanza Decor November 25, 2025
I’m a pharmacist. I’ve seen patients cry because they couldn’t afford brand-name meds. Then generics came. Cheaper. Safe. Effective. And GDUFA made that possible. I don’t care if the fees are high. I care that my patients get their meds on time. This system works. Don’t break it. Fix the edges-but don’t throw out the baby with the bathwater.